Abstract
The MIQE guidelines have set a new standard for publishing qPCR results to ensure integrity in the scientific literature and increase experimental transparency. One of the key guidelines focuses on the proper design and experimental validation of primer assays used in qPCR. This tech report describes the design and wet-lab validation of PrimePCR assays and how they comply with the MIQE guidelines.
Introduction
Quantitative PCR (qPCR) is a powerful tool in life science research. However, the data it generates can only be trusted if sufficient attention is paid to quality control and quality assurance at every step of the experimental workflow. One particular example underlines how wrong conclusions can be drawn from qPCR experiments that are not validated properly. A study was published in which an association was claimed between the presence of measles virus and gut pathology in children with developmental disorders (Uhlmann et al. 2002). These data were subsequently used to infer an association between autism and MMR vaccination. Even though independent experts revealed serious anomalies in the qPCR data and their interpretation and refuted the claimed association, a consequence of this and other reports was that measles has become endemic in the United Kingdom as the result of people refusing vaccination because of these claims. The lack of documentation of proper quality control in far too many published qPCR experiments led a team of international qPCR experts to develop the minimum information for publication of quantitative real-time PCR experiments (MIQE) guidelines (Bustin et al. 2009). Amongst others, these guidelines describe the types of validation that should be performed prior to the use of a qPCR assay. This includes both in silico analysis, such as verifying the absence of single nucleotide polymorphisms (SNPs) or secondary structures in the primer annealing regions that may result in biased or reduced amplification, and wet-lab validation of PCR efficiency and specificity.

and Jan Hellemans
Cofounders, Biogazelle
Commercial assay providers have traditionally used computer-based design pipelines for generating qPCR assays that in theory should meet quality criteria. However, this does not guarantee that the assay will actually work properly in the lab. To meet the MIQE guidelines, end users still need to perform the wet-lab validation. This situation is far from ideal because not all qPCR users are aware that those assays have not been fully validated. In addition, the assay validation by individual researchers leads to a waste of precious time and resources. Bio-Rads PrimePCR assays are a unique and attractive alternative, meeting the MIQE guidelines by thorough validation of every single assay using rigorous and state-of-the-art laboratory testing procedures.
PrimePCR Assay Design
During the design phase of the SYBR Greenbased qPCR assays for all human protein-coding genes, the following key requirements were achieved.
1. High assay specificity without use of a probe. Despite the fact that probes may increase the detection specificity of an assay, undetected coamplification of nonspecific products (off-target amplification and primer-dimers) may negatively impact the performance of the assay, even for probe-based assays. PrimePCR assays are based on state-of-the-art specificity prediction algorithms, followed by next generation sequence verification of each amplicon.
2. Compatibility with standard assay conditions. All assays should work with validated and universal PCR conditions.
3. Avoided secondary structures in primer annealing sites. Secondary DNA structures in primer annealing sites are known to prevent efficient annealing of the primer and hence reduce amplification efficiency.
4. Avoided SNPs in target regions. SNPs located in the primer binding sites (particularly the 3′ end) may interfere with proper annealing of a primer to its target sequence. This may result in impaired amplification of the variant allele, or even allele-specific amplification. Because of the sheer number of SNPs being discovered in the human transcriptome it is becoming impossible to avoid SNPs in all primers, thus necessitating the acceptance of assays with SNPs in the 5′ end of a primer, but shown experimentally to have no impact on assay performance.
5. Maximized fraction of transcript isoforms being detected. The majority of human protein-coding genes are transcribed into different messenger RNA (mRNA) isoforms. These alternative transcripts typically share significant parts of the gene but some gene regions only occur in a limited number of transcripts or even in just a single rare isoform. These regions should be avoided as they rarely yield the results researchers are interested in.
6. Designed intron-spanning assays whenever possible. Unless preventive measures are taken, RNA is typically contaminated with genomic DNA (gDNA). This DNA will consequently be present in the complementary DNA (cDNA) sample where it may lead to unwanted amplification for some assays. By designing assays spanning sufficiently large introns (proven to prevent or substantially reduce gDNA coamplification), only cDNA in which the introns have been spliced out will be amplified. This design method is not possible for all genes because some genes only have one exon or the introns are not of sufficient size to prevent amplification.
7. Used latest release of genome builds and annotation databases. The human genome is constantly curated with new information. It is therefore paramount that the latest information is used and that assay annotation is constantly updated. PrimePCR assays are based on the most recent gene annotation, SNP, and transcript variant information, and will be regularly updated as new information accumulates.
Validation
{loadposition mp3pos1} {jwplayer}http://www.bioradiations.com/media/audio/primepcr_assays_wet-lab_validation_audio.mp3&height=18&width=340{/jwplayer}
Listen to Jo Vandesompele and Jan Hellemans, cofounders of Biogazelle and validation team for the PrimePCR assays, talk about qPCR, MIQE guidelines, and their design and experimental validation process for the PrimePCR assays.
All PrimePCR assays underwent thorough wet-lab validation. In accordance with the MIQE guidelines, assays were evaluated in terms of specificity, efficiency, linear dynamic range, and background signal in negative controls. At least ten qPCR reactions were performed for each assay: cDNA from reference RNA, no-template control (NTC), gDNA, and seven points from a tenfold dilution series of synthetic templates (from 20 million down to 20 copies). All assays were tested using SsoAdvanced SYBR Green supermix (Bio-Rad Laboratories, Inc.) and 1x final primer concentration, and were run in a CFX384 Touch real-time PCR detection system (Bio-Rad) using standard cycling parameters. Data analysis was performed using CFX Manager software (Bio-Rad) with autocalculated baseline and fixed threshold settings (300 relative fluorescence units [RFU]). For the cDNA samples, qPCR human reference total RNA (Agilent Technologies, Inc.) was reverse transcribed using iScript advanced cDNA synthesis kit for RT-qPCR (Bio-Rad), and 25 ng of cDNA was used as input in a 5 l qPCR reaction. For gDNA samples, 2.5 ng of human gDNA (Roche Diagnostics) was used. Both the dilution series and NTC included 5 ng/l yeast transfer RNA (tRNA) as carrier to reduce potential issues at low template concentration.
Amplification efficiencies were calculated from the results of the dilution series. Only assays that displayed good linear performance in the 2020 million copy number range (r2 >0.99) with efficiencies between 90 and 110% were considered to be of sufficient quality (Figure 1). The average y-intercept of the standard curves was found to be 35, indicating that a single template molecule results in a quantification cycle (Cq) value of 35 when amplified with SsoAdvanced SYBR Green supermix in a qPCR reaction (Figure 2).
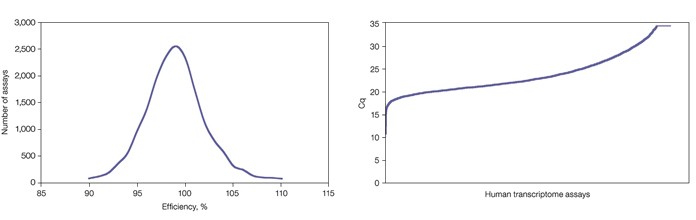
Fig. 1. Distribution of PrimePCR assay efficiencies. While efficiencies of 90110% were targeted, final analysis shows that a large majority of assays actually fall between 95 and 105%. When assays fail to meet the 90110% efficiency target, assay redesign and validation are attempted up to three times.Fig. 2. Quantification of human reference RNA revealed a wide range of expression levels across all genes. The lowest Cq value, observed for ND4L, equals 11. This results in a Cq range of 24 cycles (assuming a single transcript copy has a Cq of 35), or an observed expression range of at least 16 million-fold.
A second essential quality criterion is the specificity of the assays. Specificity is typically assessed by evaluating the size and melting temperature (Tm) of the PCR product. In qPCR, a single melt peak at the expected Tm for the amplicon is typically accepted as a good specific assay. Unfortunately, the sensitivity of these methods is too limited to detect small fractions of nonspecific PCR products generated from low-abundance transcripts. It is also known to sometimes generate more than one peak for assays that are confirmed to be specific (domains with different GC content are known to produce melt curves with more than one peak). To ensure high specificity, all PCR products from cDNA were validated by large-scale massive parallel sequencing. Only assays for which all reads mapped to the target gene were considered to be specific.
The sensitivity of qPCR reactions is mainly determined by the degree to which background signal can be avoided. Therefore, a third quality control measures the signal in an NTC reaction. If no natural template is present in the PCR reaction, the polymerase tends to amplify nonspecific products, mainly primer-dimers. If this nonspecific amplification results in a Cq value below 35 (the Cq value for a single molecule of template), the sensitivity of the assay is compromised. Therefore, all the assays with an NTC Cq value
To further strengthen the value of the PrimePCR assay validation study, two types of references were included. First, a large quantity of reverse transcribed human reference total RNA (25 ng total RNA mass equivalents) was used for all PrimePCR assays. Since this reference RNA is commercially available and the expression levels for all human protein- coding genes in this sample are made public by Bio-Rad online at www.bio-rad.com/PrimePCR, it can be used as a resource for cross-validation. Secondly, throughout the validation studies a spike-in control (SIC) template was used in every 384-well plate. In this case, and in future studies, the use of SIC allows the comparison of data among different runs. By including this universal reference (and calibration) point, data can be compared across different labs and instruments (Vermeulen et al. 2009).
Conclusions
All PrimePCR assays were designed following strict guidelines on maximum transcript coverage, minimal overlap with known SNPs, and spanning large introns where possible. In addition, they have all been fully validated in the lab, passing stringent quality controls. As a result, PrimePCR assays set a new quality standard for commercially available predesigned qPCR assays (Bustin et al. 2011). In accordance with the MIQE guidelines (Bustin et al. 2009), there is full transparency on the performance of every PrimePCR assay in the form of a standardized specification and validation sheet that can be found on the Bio-Rad website, www.bio-rad.com/PrimePCR.
References
Bustin SA et al. (2009). The MIQE guidelines: Minimum information for publication of quantitative real-time PCR experiments.
Clin Chem 55, 611622.
Bustin SA et al. (2011). Primer sequence disclosure: A clarification of the MIQE guidelines. Clin Chem 57, 919921.
Uhlmann V et al. (2002). Potential viral pathogenic mechanism for new variant inflammatory bowel disease. Mol Pathol 55, 8490.
Vermeulen J et al. (2009). External oligonucleotide standards enable cross laboratory comparison and exchange of real-time quantitative PCR data. Nucleic Acids Res 37, e138.
SYBR is a trademark of Life Technologies Corporation. Bio-Rad Laboratories, Inc. is licensed by Life Technologies Corporation to sell reagents containing SYBR Green I for use in real-time PCR, for research purposes only.
Bio-Rads real-time thermal cyclers are licensed real-time thermal cyclers under Appleras U.S. Patent Number 6,814,934 B1 for use in research, human in vitro diagnostics, and all other fields except veterinary diagnostics.
Bio-Rads real-time thermal cyclers are covered by one or more of the following U.S. patents or their foreign counterparts owned by Eppendorf AG: U.S. Patent Numbers 6,767,512 and 7,074,367.
Purchase of SsoAdvanced SYBR Green supermix includes an immunity from suit under patents specified in the product insert to use only the amount purchased for the purchasers own internal research. No other patent rights are conveyed expressly, by implication, or by estoppel. Further information on purchasing licenses may be obtained by contacting the Director of Licensing, Applied Biosystems, 850 Lincoln Centre Drive, Foster City, California 94404, USA.