When it comes to tumor profiling, the poor quality and degradation of RNA in formalin-fixed paraffin-embedded (FFPE) samples are major limitations. To overcome this challenge, we tested a novel RNA-Seq workflow using the SEQuoia Complete Library Prep Kit and the SEQuoia Ribodepletion Kit. This workflow offers more complex transcriptome profiling, including both long and short RNA biotypes, to better represent the complete transcriptome.
RNA sequencing (RNA-Seq) is a popular technique for genome-scale analysis of RNA in a sample because it is the most powerful tool available for examining the quantity and transcripts of RNA. By comparing gene transcripts to a reference genome standard, researchers can even measure absolute gene expression levels in thousands of genes under one condition or compare it across multiple conditions. Thanks to RNA-Seq, comprehensive and quantitative genomic data is now widely available, causing a paradigm shift in the life sciences. In cancer research, RNA-Seq has enabled the discovery of new biomarkers, analysis of tumor heterogeneity, screening for drug therapy resistance, and many other breakthroughs (Hong M et al. 2020).
Several key challenges currently prevent the broader use of RNA-Seq. FFPE samples are the most common archived specimen type available, making them a key resource for researchers; yet, the low yield and poor quality of degraded RNA obtained from these samples can make them unsuitable for use in RNA-Seq.
Regardless of sample type, researchers must also choose a method for library preparation. Due to the inherent bias in many commercial products, researchers must typically choose to focus on either long (>200 bp) or short (<200 bp) RNAs.
Fortunately, a recent application note shows a new approach for better transcriptome analysis of FFPE samples. In a study of breast cancer samples, RNA-Seq library preparation with SEQuoia Kits showed the ability to overcome these biases and provide quality data, including both short and long RNAs from a single workflow. Implementing the SEQuoia workflow strategy can offer researchers more complete information from their samples at lower cost and effort, and also enables access to high-quality data even if highly degraded RNA from FFPE samples is used.
Key considerations from the study are explored below.
The typical RNA-Seq workflow tends to be a lengthy, multistep process that involves sample RNA extraction, ribosomal RNA (rRNA) removal using either enrichment or depletion, chemical or enzymatic fragmentation of the residual RNA into smaller fragments, reverse transcription of RNA into smaller fragments, and finally, amplification of the library pool to yield a quantity that is sufficient to load onto a sequencing flow cell.
The degraded and heavily modified nature of FFPE RNA presents key limitations for the traditional library preparation strategy due to its high RNA quality and yield requirements, which make the workflow more complex (Palomares et al. 2019, Sarantopoulou et al. 2019).
Most RNA-Seq library preparation protocols focus on capturing only long RNA fragments unless specifically designed to isolate small RNA fragments (Naphade et al. 2022). Evidence suggesting that both small RNAs and long noncoding RNAs (lncRNAs) are key in gene expression regulation encourages the capture of both classes of RNAs in RNA-Seq libraries (Patil et al. 2014, Ransohoff et al. 2018).
However, generating data that represents the complete transcriptome requires the costly and laborious construction, sequencing, and analysis of multiple libraries. Furthermore, most library preparation strategies deplete rRNA before reverse transcription. The smaller RNA fragments that are a hallmark of FFPE RNA samples may be further degraded and lost through handling during the depletion step. In short, obtaining a complete and biologically representative transcriptomic dataset from FFPE RNA is a challenge.
A novel RNA-Seq library preparation workflow (Figure 1) has recently been introduced that aims to overcome many of the challenges associated with generating complex, rich data from FFPE-derived degraded RNA. The SEQuoia Complete Stranded RNA Library Prep Kit leverages a proprietary retrotransposon enzyme in place of the traditional reverse transcriptase and ligases found in other RNA-Seq library preparation kits. This enzyme possesses greater processivity than a retroviral reverse transcriptase and readily conducts end-to-end template jumping (Bibillo and Eickbush 2002, Bibillo and Eickbush 2004), enabling a continuous synthesis reaction that converts RNA to cDNA and adds both sequencing adapters in a single step.
By first polyadenylating the RNA fragments and then adding a polythymidine sequence to the 3′ sequencing adapter to prime the reaction, this enzyme captures long and short RNA biotypes as well as degraded RNA fragments in a single library. Using this kit in conjunction with the SEQuoia RiboDepletion Kit (for rRNA-derived fragment depletion) yields a comprehensive library from FFPE RNA that is comprised of both long and short RNA biotypes and therefore provides better representation of the complete transcriptome.
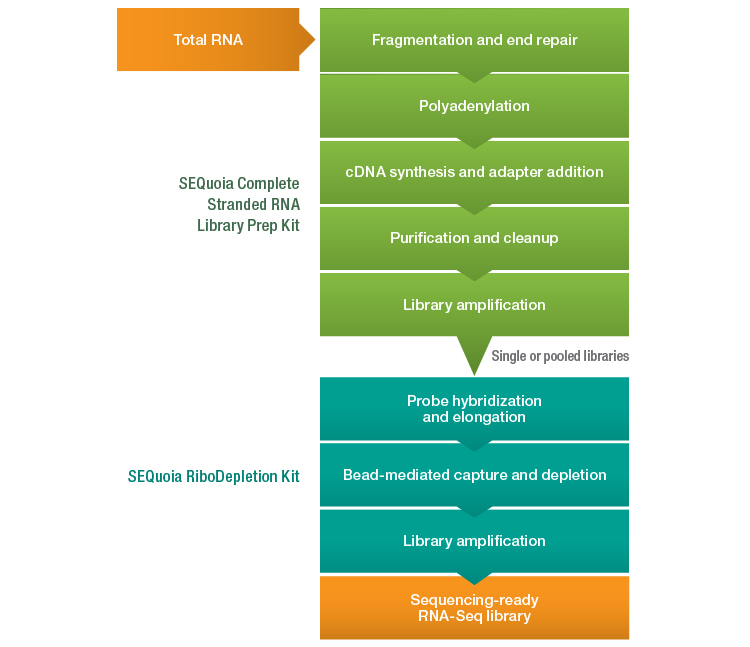
Fig. 1. Novel RNA-Seq library preparation workflow employing the SEQuoia Complete Stranded RNA Library Prep and SEQuoia RiboDepletion Kits. cDNA, complementary DNA; RNA-Seq, RNA sequencing.
Three library construction workflows were evaluated to assess the suitability of pre-library versus post-library ribodepletion and to compare the ability of different library chemistries to construct complex libraries that represent the whole transcriptome.
There are key distinctions between the RNA biotypes represented in the RNA-Seq libraries from each workflow tested (SEQ, Hybrid, NEB). Whereas the total number of genes detected was similar across the workflows and between FFPE and flash frozen (FF) samples, significantly more small RNA biotypes were captured in workflows that employed SEQuoia Complete Stranded RNA Library Prep Kit (Figure 2A). Depleting rRNA before library construction resulted in a loss of small RNA biotypes (compare SEQ to Hybrid workflow), but the SEQuoia Complete Stranded RNA Library Prep Kit still captured more small RNA biotypes that remained after depletion (compare Hybrid to NEB workflow). The capture of miRNAs by the SEQuoia Complete Library Prep Kit from total RNA was significantly more than the number of miRNAs captured by the same method using pre-depleted RNA (Figure 2B), indicating that loss of valuable material occurs through pre–library prep sample handling.
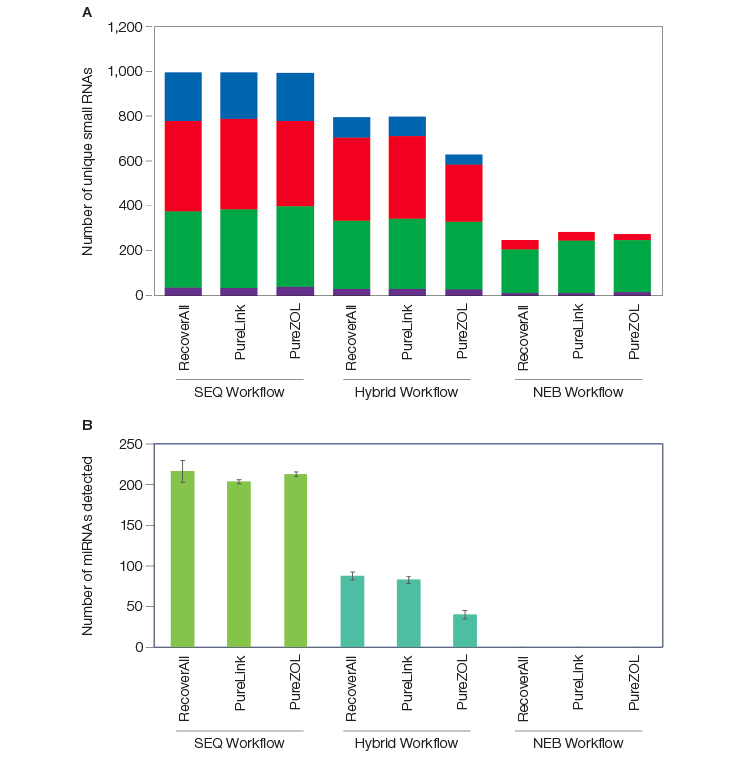
Fig. 2. More small RNAs were detected with post-library ribodepletion than with pre-library ribodepletion. The number of unique transcripts per RNA biotype detected in libraries prepared using different RNA isolation methods (Thermo Fisher RecoverAll Kit, Thermo Fisher PureLink Kit, and Bio-Rad PureZOL Reagent) and a SEQuoia Kit workflow (SEQ), a workflow using NEB NEBNext Kits (NEB), and a hybrid workflow (Hybrid) for library preparation are shown in the following panels: A, combined unique small RNAs, including miRNA (■), tRNA (■), snoRNA (■), and snRNA (■), detected with ≥5 reads each; B, unique miRNAs detected at ≥5 reads. Error bars represent standard deviation. lncRNA, long noncoding RNA; miRNA, microRNA; RPKM, reads per kilobase of transcript per million reads mapped; snRNA, small nuclear RNA; snoRNA, small nucleolar RNA; tRNA, transfer RNA.
There is a significant difference in the RNA biotypes captured by each of the workflows (Figure 3). The libraries prepared using SEQuoia Kits (SEQ workflow) were much more complex in terms of the small RNA biotypes. Notably, over 500 unique small RNAs, including miRNA, tRNA, snRNA, and snoRNA subtypes, were detected in SEQ workflow libraries (Figure 3B), compared with only seven small RNAs in the NEB workflow libraries.
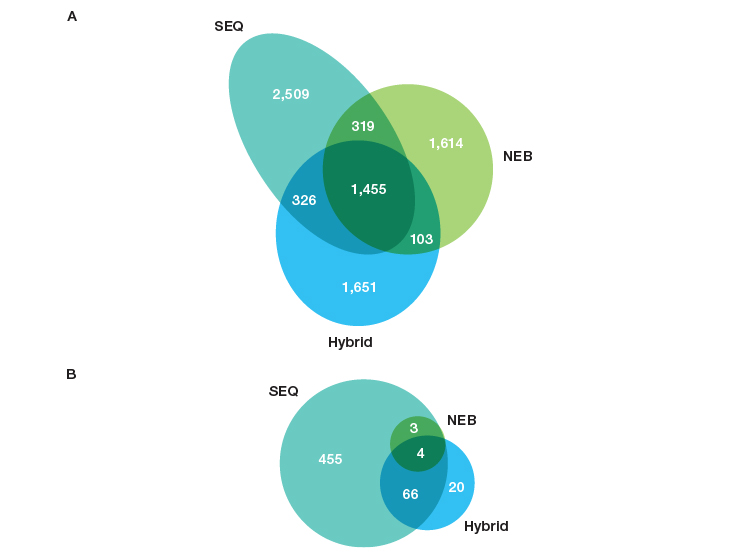
Fig. 3. SEQuoia Kit library workflow provides more complete transcriptome coverage of small RNAs in addition to long RNAs. Venn diagrams demonstrating the number of genes detected at ≥1 RPKM and ≤10% CV in libraries constructed from FFPE RNA extracted using the Thermo Fisher RecoverAll Kit to isolate RNA with the same library prep/ribodepletion. A, lncRNA and protein-coding genes; B, small RNAs (miRNA, tRNA, snoRNA, snRNA). CV, coefficient of variation; lncRNA, long noncoding RNA; miRNA, microRNA; RPKM, reads per kilobase of transcript per million reads mapped; snRNA, small nuclear RNA; snoRNA, small nucleolar RNA; tRNA, transfer RNA.
Higher throughput applications can benefit from the use of library pooling, which offers less effort and substantial cost-savings from sequencing compared to individual libraries. This study considered whether data quality would be impacted by multiplexing libraries using the SEQuoia RiboDepletion Kit.
In a separate experiment, 48 RNA-Seq libraries were constructed and indexed using SEQuoia Complete Stranded RNA Library Prep Kit. Each library was split into two; half of each library was ribodepleted individually using the SEQuoia RiboDepletion Kit, and the other half of each library was pooled by mixing an equimolar ratio of libraries and then depleted en masse using the SEQuoia RiboDepletion Kit. The percentage of reads mapping to rRNA in libraries that were depleted en masse was consistently within 5% of the same library depleted in a singleplex reaction (Figure 4A). In addition, the transcriptome profile is preserved when libraries are depleted en masse, as illustrated in Figure 4B, where the Pearson correlation coefficient of transcripts between the multiplexed and single ribodepletion reactions ranged from 0.977 to 0.996, with an average of 0.992.
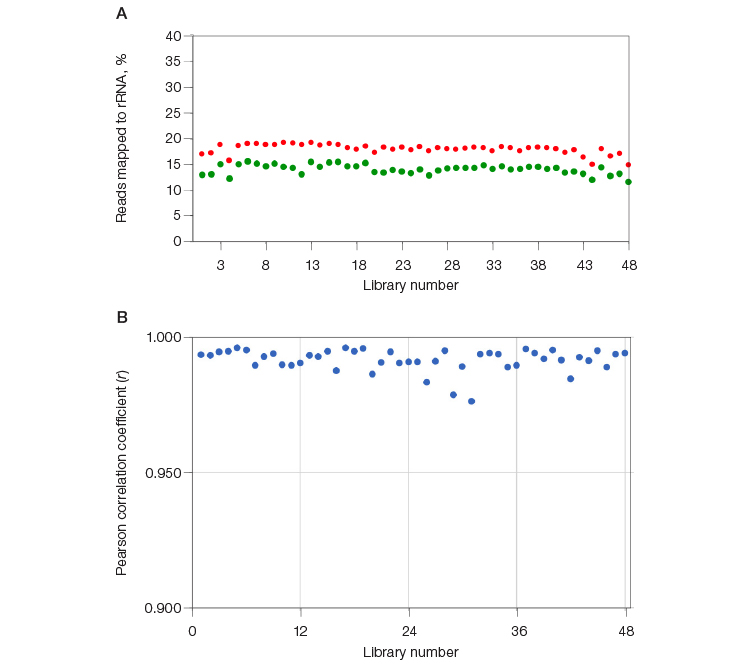
Fig. 4. Multiplexing capability of SEQuoia RiboDepletion Kit. Forty-eight libraries were constructed using the SEQuoia Complete Stranded RNA Library Prep Kit. An aliquot of each library was either individually depleted using the SEQuoia RiboDepletion Kit or pooled by mixing an equal molar ratio of the libraries and then depleted. A, the percentage of reads mapped to rRNA for each library are plotted (⬤, multiplexed depletion; ⬤, singleplex depletion). B, the number of total unique transcripts with RPKM > 0.1 in the libraries depleted in the pooled reaction was compared to the number in the individually depleted libraries. The Pearson correlation coefficient was calculated between the individually and multiplexed ribodepleted libraries. RPKM, reads per kilobase of transcript per million reads mapped; rRNA, ribosomal RNA.
Conclusion
Overall, these data show that the SEQuoia Complete RNA Library Prep Kit and the SEQuoia RiboDepletion Kit enable more complete capture of the transcriptome for RNA-Seq and are suitable for use with FFPE samples. Samples can also be pooled for cost savings and higher throughput without sacrificing data quality.
To read more about this study, check out the application note, Comprehensive Capture of FFPE RNA for RNA-Seq Using a Continuous Synthesis Chemistry and Post-Library Ribodepletion.
References
Hong M et al. (2020). RNA sequencing: new technologies and applications in cancer research. J Hematol Oncol 13, 166.
Bibillo A and Eickbush TH (2002). High processivity of the reverse transcriptase from a non-long terminal repeat retrotransposon. J Biol Chem 277, 34,836–34,845.
Bibillo A and Eickbush TH (2004). End-to-end template jumping by the reverse transcriptase encoded by the R2 retrotransposon. J Biol Chem 279, 14,945–14,953.
Naphade S et al. (2022). Systematic comparative analysis of strand-specific RNA-seq library preparation methods for low input samples. Sci Rep 12, 1789.
Palomares MA et al. (2019). Systematic analysis of TruSeq, SMARTer and SMARTer Ultra-Low RNA-Seq kits for standard, low and ultra-low quantity samples. Sci Rep 9, 7550.
Patil VS et al. (2014). Gene regulation by non-coding RNAs. Crit Rev Biochem Mol Biol 49, 16–32.
Ransohoff JD et al. (2018). The functions and unique features of long intergenic non-coding RNA. Nat Rev Mol Cell Biol 19, 143–157.
Ribarska T et al. (2022). Optimization of enzymatic fragmentation is crucial to maximize genome coverage: A comparison of library preparation methods for Illumina sequencing. BMC Genomics 23, 92.
Sarantopoulou D et al. (2019). Comparative evaluation of RNA-Seq library preparation methods for strand-specificity and low input. Sci Rep 9, 13477.
van Schooneveld E et al. (2015). Dysregulation of microRNAs in breast cancer and their potential role as prognostic and predictive biomarkers in patient management. Breast Cancer Res 17, 21.


